The fastq sequence format¶
Note
This article uses material from the Wikipedia article FASTQ format <http://en.wikipedia.org/wiki/FASTQ_format> which is released under the Creative Commons Attribution-Share-Alike License 3.0 <http://creativecommons.org/licenses/by-sa/3.0/>
fastq format is a text-based format for storing both a biological sequence (usually nucleotide sequence) and its corresponding quality scores. Both the sequence letter and quality score are encoded with a single ASCII character for brevity. It was originally developed at the Wellcome Trust Sanger Institute to bundle a The fasta format sequence and its quality data, but has recently become the de facto standard for storing the output of high throughput sequencing instruments such as the Illumina Genome Analyzer Illumina. 1
Format¶
A fastq file normally uses four lines per sequence.
Line 1 begins with a ‘@’ character and is followed by a sequence identifier and an optional description (like a The fasta format title line).
Line 2 is the raw sequence letters.
Line 3 begins with a ‘+’ character and is optionally followed by the same sequence identifier (and any description) again.
Line 4 encodes the quality values for the sequence in Line 2, and must contain the same number of symbols as letters in the sequence.
A fastq file containing a single sequence might look like this:
@SEQ_ID
GATTTGGGGTTCAAAGCAGTATCGATCAAATAGTAAATCCATTTGTTCAACTCACAGTTT
+
!''*((((***+))%%%++)(%%%%).1***-+*''))**55CCF>>>>>>CCCCCCC65
The character ‘!’ represents the lowest quality while ‘~’ is the highest. Here are the quality value characters in left-to-right increasing order of quality (ASCII):
!"#$%&'()*+,-./0123456789:;<=>?@ABCDEFGHIJKLMNOPQRSTUVWXYZ[\]^_`abcdefghijklmnopqrstuvwxyz{|}~
The original Sanger FASTQ files also allowed the sequence and quality strings to be wrapped (split over multiple lines), but this is generally discouraged as it can make parsing complicated due to the unfortunate choice of “@” and “+” as markers (these characters can also occur in the quality string).
Variations¶
Quality¶
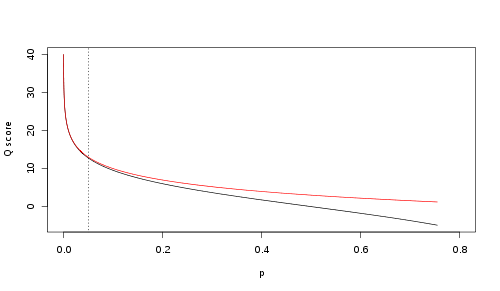
A quality value Q is an integer mapping of p (i.e., the probability that the corresponding base call is incorrect). Two different equations have been in use. The first is the standard Sanger variant to assess reliability of a base call, otherwise known as Phred quality score:
\(Q_\text{sanger} = -10 \, \log_{10} p\)
The Solexa pipeline (i.e., the software delivered with the Illumina Genome Analyzer) earlier used a different mapping, encoding the odds p/(1-p) instead of the probability p:
\(Q_\text{solexa-prior to v.1.3} = -10 \, \log_{10} \frac{p}{1-p}\)
Although both mappings are asymptotically identical at higher quality values, they differ at lower quality levels (i.e., approximately p > 0.05, or equivalently, Q < 13).
Encoding¶
Sanger format can encode a Phred quality score from 0 to 93 using ASCII 33 to 126 (although in raw read data the Phred quality score rarely exceeds 60, higher scores are possible in assemblies or read maps).
Solexa/Illumina 1.0 format can encode a Solexa/Illumina quality score from -5 to 62 using ASCII 59 to 126 (although in raw read data Solexa scores from -5 to 40 only are expected)
Starting with Illumina 1.3 and before Illumina 1.8, the format encoded a Phred quality score from 0 to 62 using ASCII 64 to 126 (although in raw read data Phred scores from 0 to 40 only are expected).
Starting in Illumina 1.5 and before Illumina 1.8, the Phred scores 0 to 2 have a slightly different meaning. The values 0 and 1 are no longer used and the value 2, encoded by ASCII 66 “B”.
Sequencing Control Software, Version 2.6, Catalog # SY-960-2601, Part # 15009921 Rev. A, November 2009]http://watson.nci.nih.gov/solexa/Using_SCSv2.6_15009921_A.pdf(page 30) states the following: If a read ends with a segment of mostly low quality (Q15 or below), then all of the quality values in the segment are replaced with a value of 2 (encoded as the letter B in Illumina’s text-based encoding of quality scores)… This Q2 indicator does not predict a specific error rate, but rather indicates that a specific final portion of the read should not be used in further analyses. Also, the quality score encoded as “B” letter may occur internally within reads at least as late as pipeline version 1.6, as shown in the following example:
@HWI-EAS209_0006_FC706VJ:5:58:5894:21141#ATCACG/1
TTAATTGGTAAATAAATCTCCTAATAGCTTAGATNTTACCTTNNNNNNNNNNTAGTTTCTTGAGATTTGTTGGGGGAGACATTTTTGTGATTGCCTTGAT
+HWI-EAS209_0006_FC706VJ:5:58:5894:21141#ATCACG/1
efcfffffcfeefffcffffffddf`feed]`]_Ba_^__[YBBBBBBBBBBRTT\]][]dddd`ddd^dddadd^BBBBBBBBBBBBBBBBBBBBBBBB
An alternative interpretation of this ASCII encoding has been proposed. Also, in Illumina runs using PhiX controls, the character ‘B’ was observed to represent an “unknown quality score”. The error rate of ‘B’ reads was roughly 3 phred scores lower the mean observed score of a given run.
Starting in Illumina 1.8, the quality scores have basically returned to the use of the Sanger format (Phred+33).
File extension¶
There is no standard file extension for a FASTQ file, but .fq and .fastq, are commonly used.