The fillgaps command¶
At the end of the oa buildgraph command the assembling graph is not always complete. Because of the non homogeneous coverage, some parts of the genome too low covered were not able to be assembled using the heuristics parameters used to assemble the main parts of the genome. The fillgaps command aims to rerun the fillgap algorithm used by the oa buildgraph but with other parameters allowing to fill the gaps not assembled during the initial assembling.
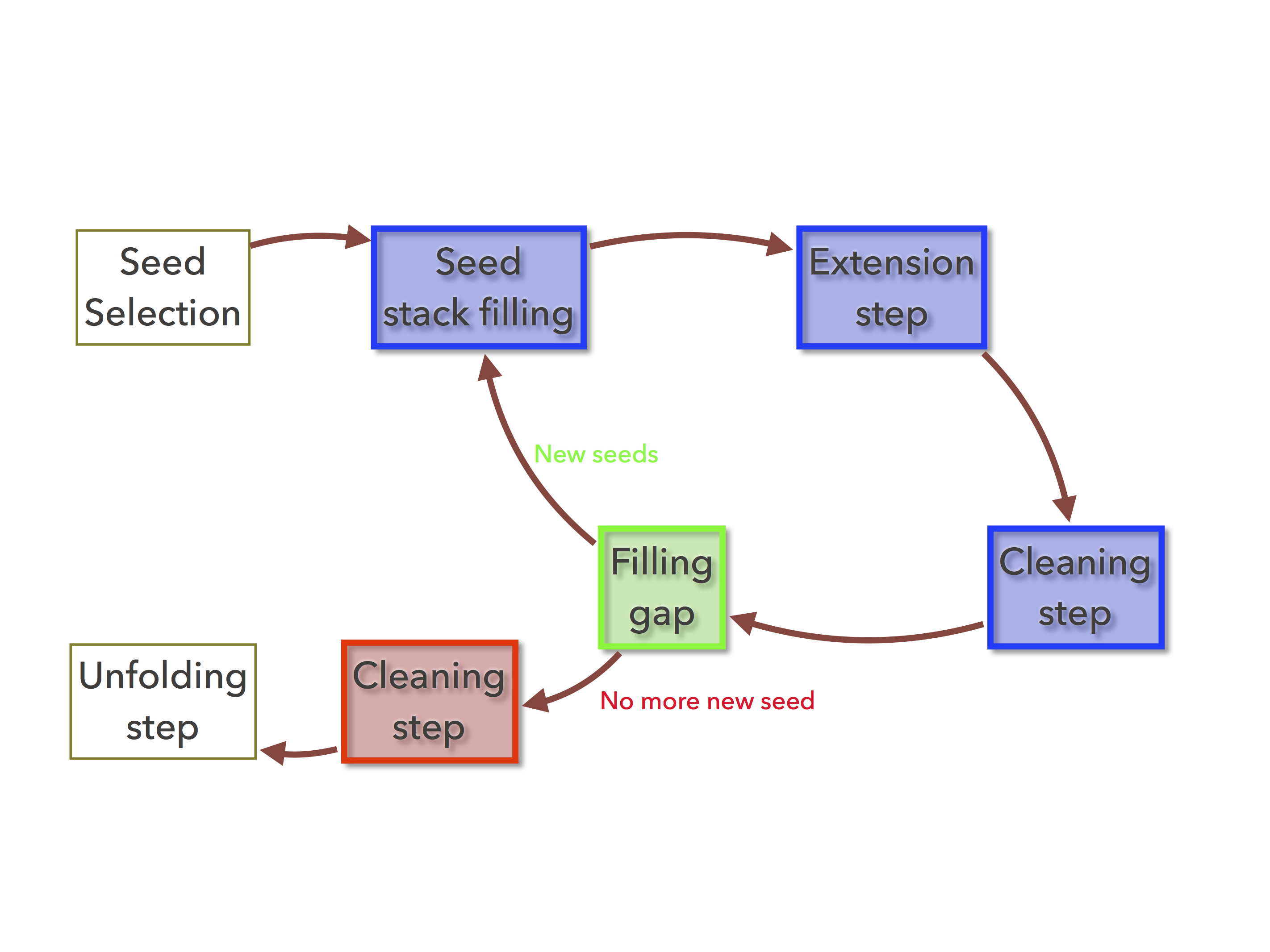
Figure 1: The organelle assembler’s fillgaps command executes all the colored tasks, starting by the green one and ending at the red task¶
command prototype¶
positional arguments¶
-
index¶ index root filename (produced by the oa index command)
-
output¶ output prefix
optional arguments¶
Graph initialisation options¶
$ oa fillgaps --seeds protChloroArabidopsis seqindex
A set of seed sequences must be or nucleic or proteic. For initiating
assembling with both nucleic and proteic sequences you must use at least two
--seeds options one for each class of sequences.
$ oa fillgaps --seeds protChloroArabidopsis --seeds rDNAChloro.fasta seqindex
-
--kupORGASM:KUP¶ The word size used to identify the seed reads [default: protein=4, DNA=12].
Graph extension options¶
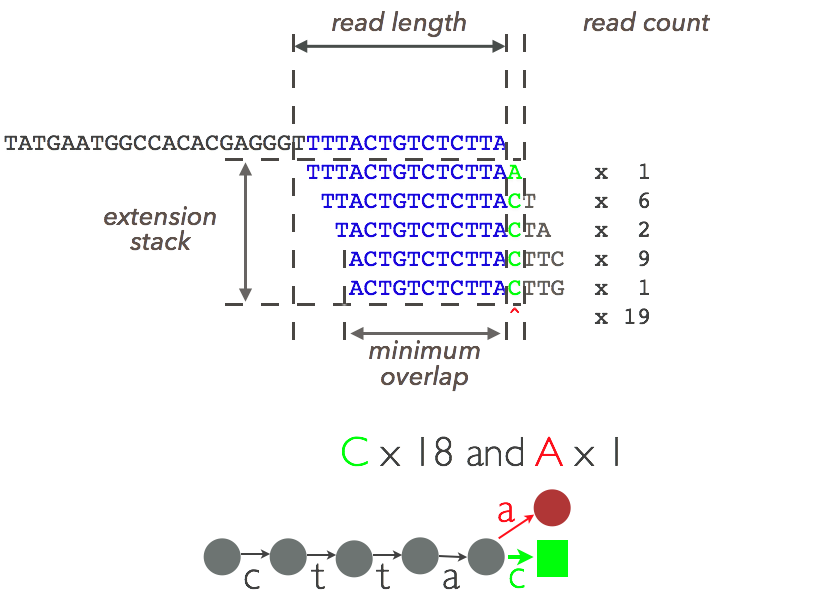
Figure 2: The assembling stack¶
-
--minreadBUILDGRAPH:MINREAD¶ the minimum count of read to consider [default: <estimated>]
$ oa fillgaps --minread 5 seqindexConsider an extension if at least five reads are present in the extension stack.
-
--coverageBUILDGRAPH:COVERAGE¶ the expected sequencing coverage [default: <estimated>]
-
--minoverlapBUILDGRAPH:MINOVERLAP¶ minimum length of the overlap between the sequence and reads to participate in the extension. [default: <estimated>]
-
--minratioBUILDGRAPH:MINRATIO¶ minimum ratio between occurrences of an extension and the occurrences of the most frequent extension to keep it. [default: <estimated>]
-
--mincovBUILDGRAPH:MINCOV¶ minimum occurrences of an extension to keep it. [default: 1]
Graph filtering options¶
-
--lowcomplexity¶ Use also low complexity probes
-
--adapt5adapt5¶ adapter sequences used to filter reads beginning by such sequences; either a fasta file containing adapter sequences or internal set of adapter sequences among [‘adapt5ILLUMINA’] [default: adapt5ILLUMINA]
-
--adapt3adapt3¶ adapter sequences used to filter reads ending by such sequences; either a fasta file containing adapter sequences or internal set of adapter sequences among [‘adapt3ILLUMINA’] [default: adapt3ILLUMINA]
Graph cleaning options¶
-
--smallbranchesBUILDGRAPH:SMALLBRANCHES¶ After a cycle a extension, if you observe the assembling graph you can observe a main path and many small aborted branches surrounding this main path. They correspond to path initiated by a sequencing error or a nuclear copy of a chloroplast region not enough covered by the skimming sequencing to be successfuly extended. One of the cleaning step consist in deleting these small branches. This option indicates up to which lenght branches have to be deleted. By default this legth is automaticaly estimated from the graph.
$ oa buildgraph --seeds protChloroArabidopsis \ --smallbranches 15 seqindex
During the cleaning steps, all the branches with a legth shorter or equal to 15 base pairs will be deleted
-
--snp¶ When the data set correspond to a pool of individuals, it is possible that natural polymorphisms artificially complexy the assembling graph. For helping the assembling process of such data set, this option will clear the graph for such SNP by keeping only the most abundant allele prsent in the dataset. The generated sequence can be considered as a king of consensus. Read can be remapped in a second time on this consensus using classical sofware like BWA to get the lost SNP information.
By default this option is deactivated
$ oa fillgaps --seeds protChloroArabidopsis \ --snp seqindexRun the assembling, ignoring the SNPs.